Workflow description
The StemCNV-check analysis workflow is designed to process Illumina SNP array data (i.e. GSA array) from raw idat files to a final report with CNV calls and SNP/SNV analysis. The workflow is implemented in snakemake and can be run with a single command once the sample table and config files are prepared and the array specific static data is generated.
- The main steps of the workflow are:
Raw data processing
Conversion of idat to gtc and vcf
SNP probe filtering
SNP annotation
CNV calling and processing
CNV calling with PennCNV and CBS
Merging of CNV calls and comparison to reference samples
Annotation of CNVs, including Check-Score calculation
CNV labelling
SNP / SNV analysis
SNV comparison to reference samples
SNV filtering to protein affecting variants and labelling
Sample identity check based on SNP genotypes
Result summary
Report generation
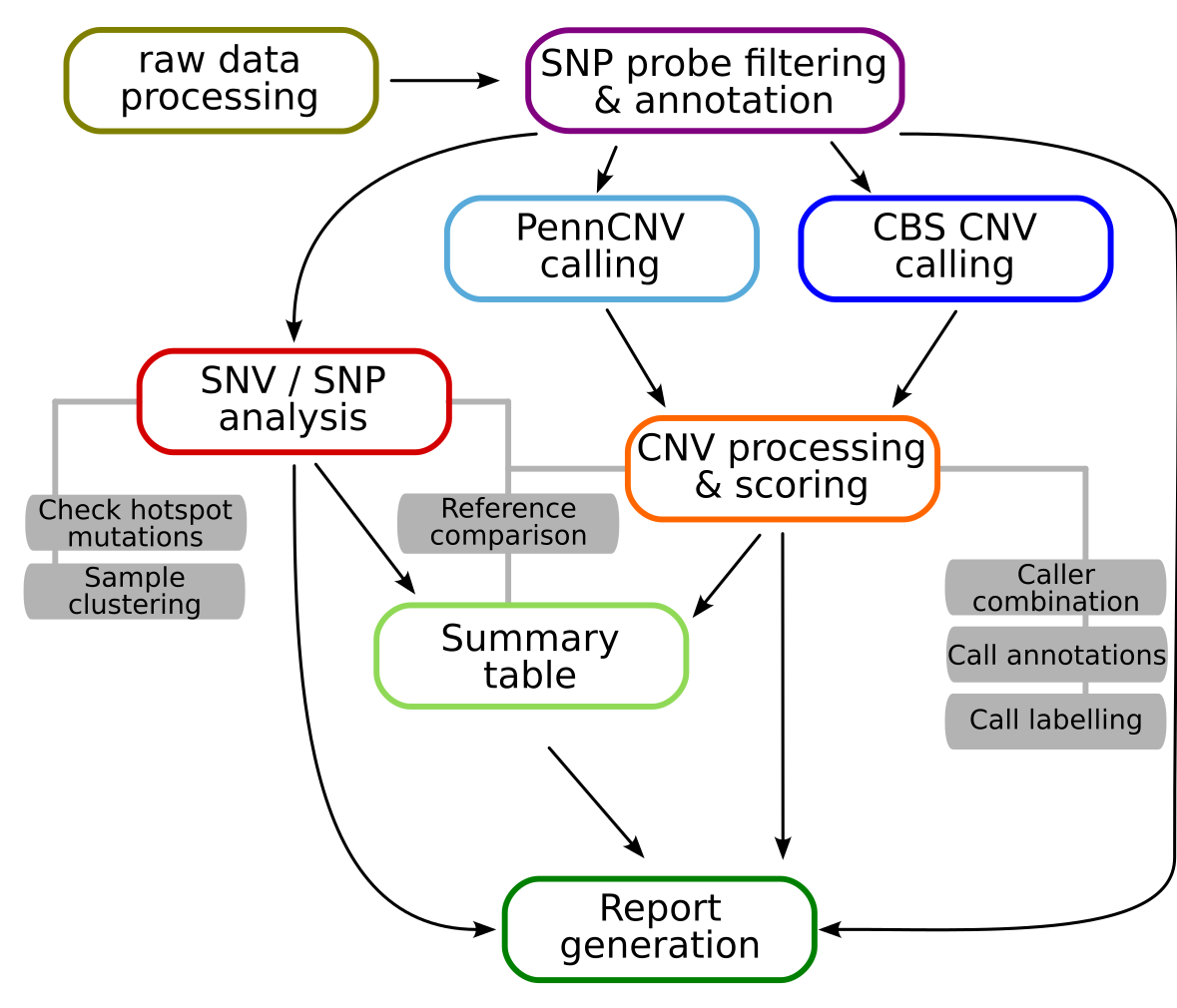
Simplified graphic overview of the StemCNV-check workflow for a single sample
The raw data processing step includes the conversion of raw idat files into the gtc format and then into vcf files. These vcf files are then extended with SNP probe filters to remove low quality probes and probes with known issues and with consequence annotations from mehari to enable later SNV analysis.
CNV calling is performed with two different algorithms, PennCNV and CBS (circular binary segmentation). The resulting CNV calls are then further processed, including merging and compared to a reference sample (if one is defined for a given sample), annotation with CNV call flags, gene overlaps, and Check-Score, and finally labelling. CNV flagging system and Check-Score will be described in an upcoming publication.
The SNP / SNV analysis includes the comparison of SNP genotypes to allow sample identity checks. Additionally, all protein changing SNVs are also compared to a reference sample (if defined) and labelled based on potential relevance.
Finally, a result summary is created for each sample and all results are collected and visualised in an easily readable html report.